



罕见病(rare disease),通常被定义为发病率低于一定比例的疾病。在不同的国家和地区,对于罕见病的定义可能有所不同。例如,在美国,罕见病被定义为影响少于20万人口的疾病,而在欧盟,罕见病被定义为影响少于2000人中的一种疾病。罕见病的特点包括:患病人数少,由于罕见病患者人数较少,许多罕见病没有得到足够的关注和研究。诊断困难,由于缺乏了解和专业知识,罕见病的诊断通常比较困难,有时需要很长时间才能确诊。治疗方案有限,很多罕见病没有有效的治疗方法,患者只能通过对症治疗来缓解症状。许多罕见病具有遗传性,常常在家族中传递。
一些常见的罕见病包括:戈谢病,一种遗传性代谢疾病,主要影响肝脏、脾脏和骨髓。亨廷顿病,一种影响大脑神经元的遗传性神经退行性疾病。肌萎缩侧索硬化症(ALS),一种影响神经系统的罕见病,导致肌肉无力和萎缩。
弗里德希氏共济失调症(Friedreich's ataxia,FRDA)是一种罕见病。作为一种遗传性神经退行性疾病,弗里德希氏共济失调症主要影响神经系统和肌肉。该疾病是一种多系统疾病,有多种症状:
运动共济失调:患者表现出步态不稳、协调性差和运动笨拙,随着病情进展,手部和手指的精细运动也会受到影响。
肌肉无力和痉挛:下肢肌肉无力和痉挛是常见症状,可能导致步态异常和行走困难。
感觉丧失:主要影响脚部和腿部的深感觉,导致平衡感减弱。
心脏问题:包括心肌病、心律失常和心脏扩大。
脊柱侧弯:许多患者会发展出脊柱侧弯(脊柱弯曲),需要矫正手术。
糖尿病:约10-20%的患者会患上糖尿病。
弗里德希氏共济失调症是一种常染色体隐性遗传疾病,由FXN基因突变引起。FXN基因负责生产弗塔汀蛋白(Frataxin),这种线粒体蛋白在能量产生和铁代谢中起重要作用。FXN基因突变导致身体缺乏Frataxin蛋白,从而影响线粒体功能,一方面造成细胞缺乏能量,另一方面抗氧化能力缺失导致损伤DNA等,最后导致细胞凋亡。大多数患者(>95%)是FXN基因homozygous,第一个内含子中 GAA 三核苷酸重复扩增,导致 转录减少和Frataxin蛋白合成减少。正常人的GAA重复次数为 7 至 40 次,FRDA患者的重复次数为 66 至 >1700 次。较少的患者在一个等位基因中发生点突变,而在另一个等位基因中 GAA 重复增加。

诊断
基因检测:检测FXN基因突变是确诊FA的金标准。
神经系统检查:评估共济失调、肌肉无力、感觉丧失等症状。
影像学检查:如MRI,检查脑部和脊髓的结构变化。
心脏检查:包括心电图和超声心动图,用于评估心脏功能。
治疗
目前,弗里德希氏共济失调症尚无治愈方法,治疗主要集中在症状管理和生活质量的改善:
药物治疗:如心脏问题的药物、糖尿病的管理药物。
物理治疗:通过运动和物理疗法来改善平衡和协调性。
外科手术:如脊柱侧弯的矫正手术。
心理支持:提供心理咨询和支持,帮助患者应对心理和情感上的挑战。
目前,科学家们正在研究多种潜在的治疗方法,包括基因治疗、药物治疗和干细胞治疗。这些研究为未来找到治愈或更有效的治疗方法带来了希望。
2023年,FDA批准Skyclarys(Omaveloxolone)用于治疗弗里德希氏共济失调症。Omaveloxolone是一种核因子E2相关因子2(Nuclear factor erythroid 2-related factor 2,Nrf2)通路激活剂。Nrf2是一种转录因子,在细胞抗氧化反应和保护细胞免受氧化应激方面起重要作用。FRDA患者由于FXN基因突变,导致弗塔汀蛋白水平降低,引起线粒体功能障碍和氧化应激增加。Omaveloxolone通过激活Nrf2通路,帮助减轻氧化应激和线粒体损伤,从而改善神经功能和缓解症状。
Nomlabofusp (CTI-1601) 是 Larimar Therapeutics 正在开发的用于治疗弗里德赖希共济失调的试验性药物。这是一种蛋白质替代疗法,用于将 frataxin 输送到细胞线粒体中,缺啥补啥。这个药在Frataxin上面加了一段CPP(Cell Penetrating Peptide,细胞穿透肽),以达到进入细胞线粒体中的目的。
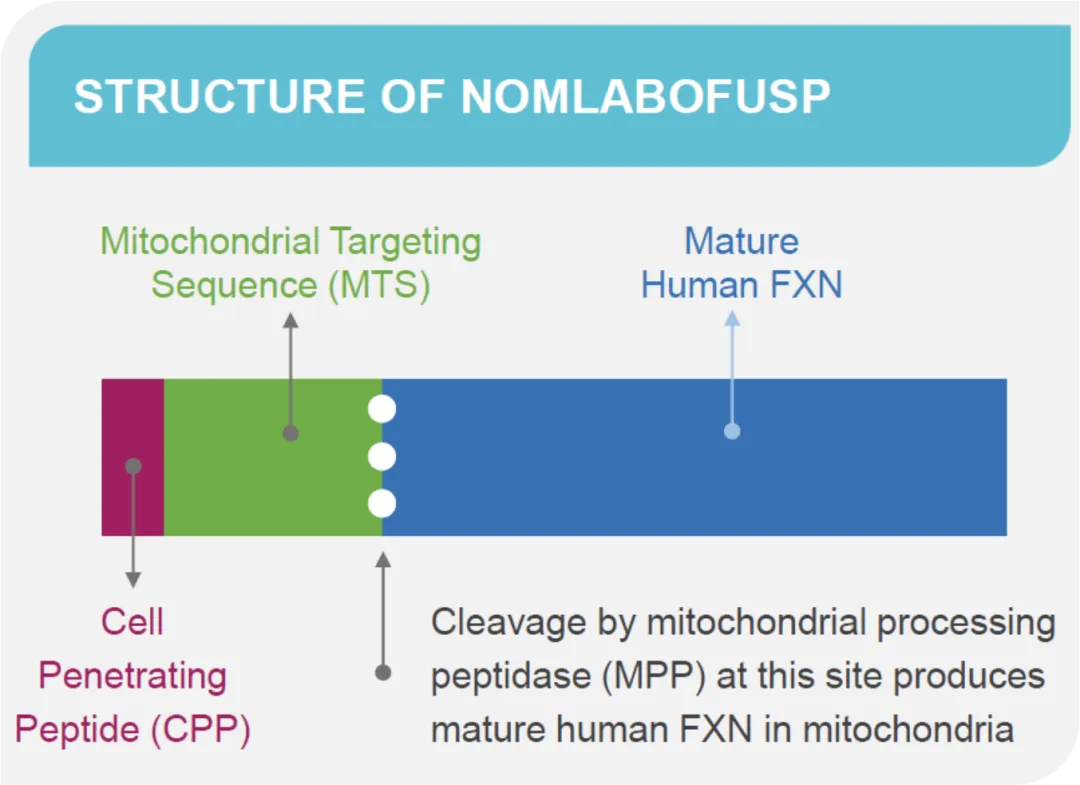
Nomlabofusp 已进行 临床2 期剂量探索试验,结果显示患者皮肤和颊细胞中 frataxin 水平增加,效果良好。FDA 最初于 2021 年因动物研究的安全问题对该药物进行了临床暂停,但这一暂停于 2022 年部分解除,并于 2023 年完全取消,允许试验进行到更高剂量组(Daily 50mg)。
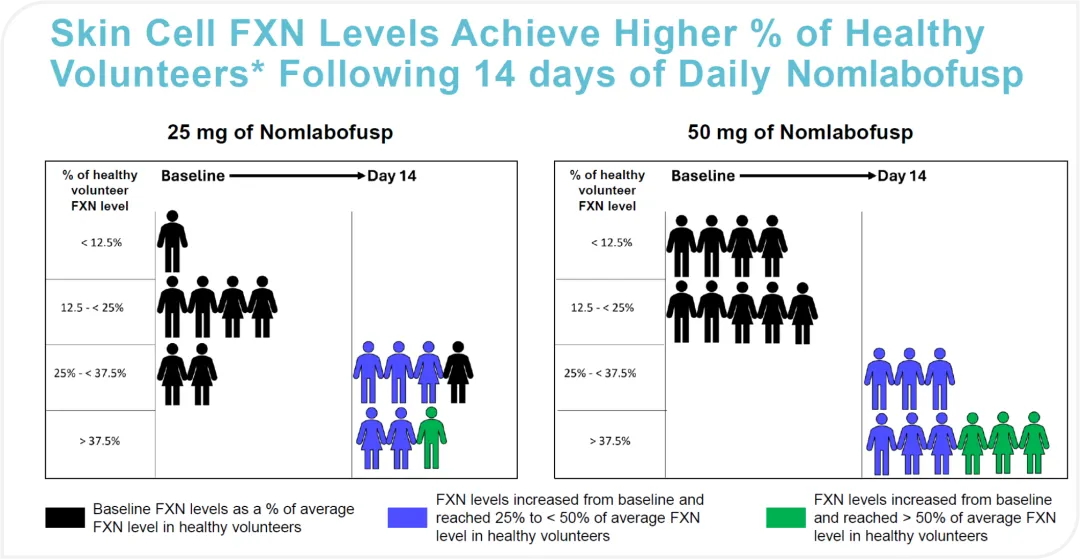
正在进行的临床开发包括一项开放标签扩展 (OLE) 研究,以评估 nomlabofusp 的长期安全性和有效性。这项研究将为未来的试验和潜在的批准申请提供剂量和给药方案。Larimar 计划2025 年下半年向FDA提交生物制品许可申请 (BLA)的申请。
